


Disease Overview
hATTR amyloidosis is a multisystem, rapidly progressive, often fatal disease1-3
Hereditary transthyretin-mediated (hATTR)
amyloidosis is an autosomal dominant disease
caused by one of many possible variants in the transthyretin (TTR) gene.3 An estimated 50,000 people are living with hATTR amyloidosis worldwide.4,5
How hATTR amyloidosis develops
Formation of amyloid deposits1,2,6

TTR TETRAMERS
TTR is primarily synthesized in the liver and is secreted as a tetramer.

TTR MONOMERS
In hATTR amyloidosis, a genetic variant causes the tetramer to become less stable, resulting in dissociation into monomers.

MISFOLDED TTR
TTR monomers misfold and aggregate into amyloid deposits.

AMYLOID DEPOSITS
Amyloid is deposited
at multiple sites in the body, causing damage that leads to clinical symptoms.

PROGRESSIVE SYMPTOMS
Accumulation of amyloid deposits over time results in worsening clinical symptoms.
Another type of ATTR amyloidosis is called wild-type ATTR amyloidosis. The etiology is unknown, but is presumed to be associated with aging.1,7
The multisystem nature of hATTR amyloidosis takes a toll on the whole body1
Multisystem involvement and family history are red flags of hATTR amyloidosis and require urgent action.3 If your patient’s family has a history of the disease
or of its many symptoms, or your patient is experiencing a combination of multisystem symptoms, the cause could be hATTR amyloidosis.



Sensory-motor neuropathy1,3
-
Length-dependent neuropathic pain and numbness
-
Altered sensation
-
Weakness
-
Difficulty walking
-
Bilateral carpal tunnel syndrome
Autonomic neuropathy1,3
-
Orthostatic hypotension
-
Diarrhea, constipation, nausea and vomiting
-
Unintentional weight loss
-
Recurrent urinary tract infections
-
Sexual dysfunction
Cardiac manifestations3,8
-
Conduction abnormalities
-
Arrhythmias
-
Heart failure
-
Left ventricular hypertrophy
Additional findings3,8
-
Family history of hATTR amyloidosis symptoms or diagnosis
-
Rapid symptom progression
-
Failure to respond to immunomodulatory treatment
-
Intolerance of commonly used cardiovascular medications
Progression of polyneuropathy leads to significant disability22



Progression of sensory-motor neuropathy1,10-121,9-11
-
Sensory loss can reduce dexterity and temperature sensation
-
Motor deficits result in progressive weakness and impaired ambulation
-
Sensory-motor neuropathy can progress more than 10x faster than diabetic neuropathy
Progression of autonomic neuropathy11-1910-19
-
Orthostatic hypotension can cause syncope and sudden falls
-
Gastrointestinal issues can cause patients to isolate themselves from social situations
-
Continuous weight loss
leads to wasting and
reduced survival -
Autonomic neuropathy can induce fatal arrhythmias
Progression of cardiac manifestations8,20-228,20-22
-
Significant and measurable decline in cardiac function results in heart failure
-
Heart failure due to hATTR amyloidosis progresses
more quickly than with other cardiac conditions
Not a comprehensive list of all the symptoms associated with hATTR amyloidosis.
Each patient may not experience all of these symptoms or may not experience them at the same time.

Multisystem dysfunction is a reality for most patients1
Although there is some association between variant and symptom presentation, most patients suffer from overlapping symptoms of sensory-motor neuropathy, autonomic neuropathy, and cardiac manifestations.1 In the United States, the most common variants (V122I, T60A, and V30M) are associated with both polyneuropathy and cardiomyopathy.1,19,23,241,18,23,24 Polyneuropathy may precede or coincide with cardiomyopathy, even in patients with the V122I variant.25
Symptom presentation of common variants in the United States13,a,b12,a,b
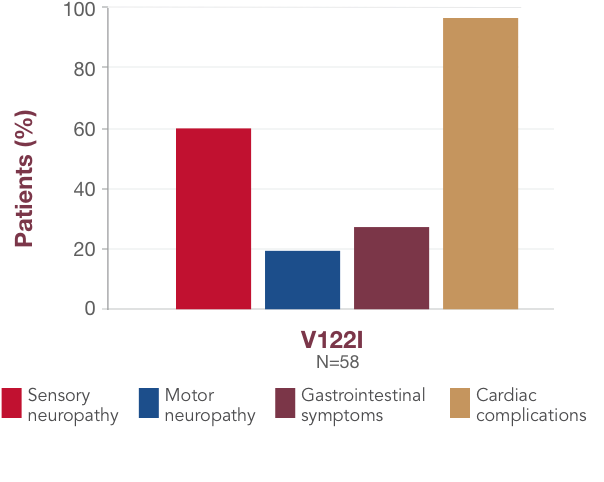
What you should know about V122I
-
V122I is the most common variant in the United States and is prevalent in ~4% of African Americans1,23,26
-
A majority of individuals with a V122I variant have polyneuropathy symptoms, including sensory, motor, and gastrointestinal symptoms12,23,27
-
In a global registry of patients with ATTR amyloidosis, 60% with the V122I variant had sensory neuropathy1312
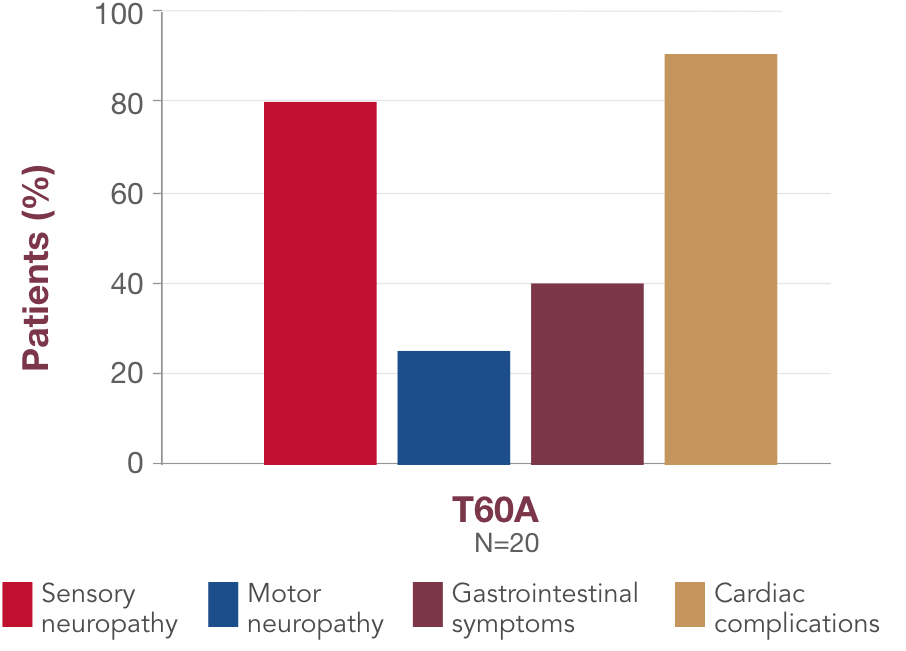
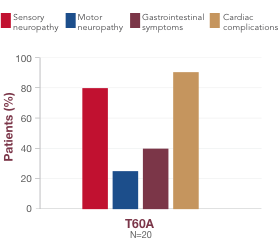
What you should know about T60A
-
T60A is the second most common variant in the US and is typically found in individuals of Irish descent23,28
-
It is associated with a mixed presentation of cardiac manifestations, autonomic neuropathy, and sensory neuropathy, and the median age of onset is 63 years13,2912,29
-
In a global registry of patients with ATTR amyloidosis, 91% of patients with the T60A variant had cardiac complications, and 80% had sensory neuropathy13,2912,29
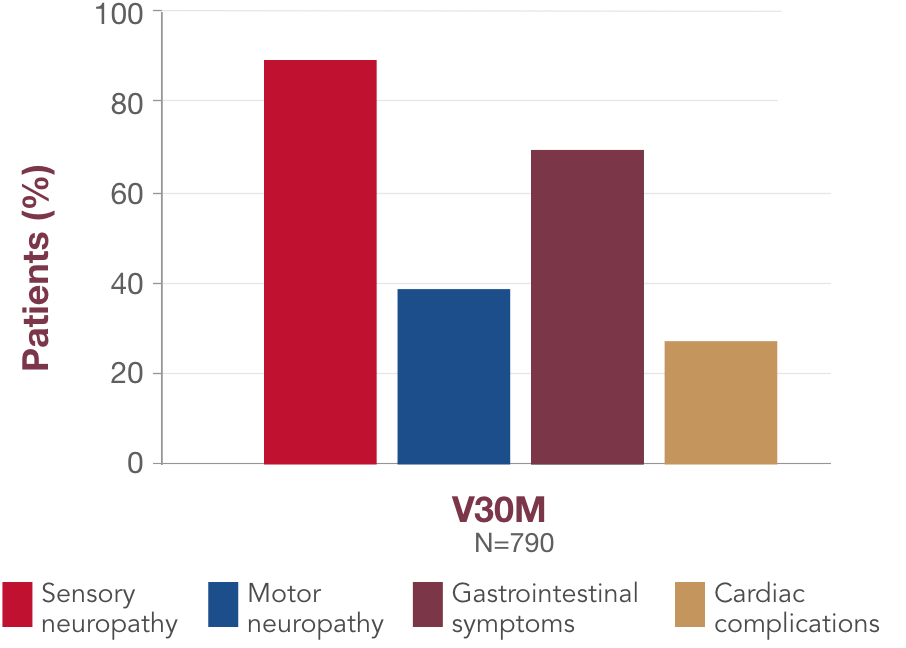
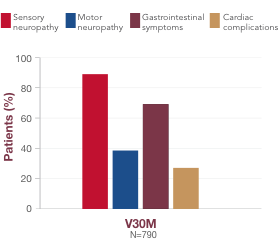
What you should know about V30M
-
V30M is the most common variant in the world, and is associated with 2 distinct clinical presentations1:
-
Early-onset V30M (<50 years) is characterized by progressive sensory-motor and autonomic neuropathy1,30
-
Late-onset V30M (≥50 years) is associated with sensory-motor neuropathy that begins in the lower limbs, with mild or no autonomic neuropathy, and has a more rapid disease course1,30
-
bData collected by the THAOS registry.
THAOS=Transthyretin-Associated Amyloidosis Outcomes Survey.
bData collected by the THAOS registry.
THAOS=Transthyretin-Associated Amyloidosis Outcomes Survey.
Patients with hATTR amyloidosis may already be in your practice
In the profiles below, you will find examples of
different patient types, which may help you to
recognize patients in your practice who may
be at
risk of hATTR amyloidosis. Patient profiles are composites created through a review of published literature and are not of actual patients.




Patient Resources


Family Resources

Information and Support Groups




References:
- Ando Y, Coelho T, Berk JL, et al. Orphanet J Rare Dis. 2013;8:31.
- Adams D, Coelho T, Obici L, et al. Neurology. 2015;85(8):675-682.
- Conceição I, González-Duarte A, Obici L, et al. J Peripher Nerv
Syst. 2016;21(1):5-9. - Plante-Bordeneuve V. J Neurol. 2014;261(6):1227-1233.
- Hawkins PN, Ando Y, Dispenzeri A, et al. Ann Med.
2015;47(8):625-638. - Kourelis TV, Gertz MA. Expert Rev Cardiovasc Ther.
2015;13(8):945-961. - Gertz MA. Am J Manag Care. 2017;23(suppl 7):S107-S112.
- Dharmarajan K, Maurer MS. J Am Geriatr Soc. 2012;60(4):765-774.
- Berk JL, Suhr OB, Obici L, et al. JAMA. 2013;310(24):2658-2667.
- Shin SC, Robinson-Papp J. Mt Sinai J Med. 2012;79(6):733-748.
- Koike H, Tanaka F, Hashimoto R, et al. J Neurol Neurosurg
Psychiatry. 2012;83(2):152-158. - Wixner J, Mundayat R, Karayal ON, et al. Orphanet J Rare Dis. 2014;9:61.
- González-Duarte A, Berk JL, Quan D, et al. J Neurol. 2020;267(3):703-712.
- González-Duarte A. Clin Auton Res. 2019;29(2):245-251.
- Ando Y, Suhr OB. Amyloid. 1998;5(4):288-300.
- Suhr O, Danielsson A, Holmgren G, et al. J Intern Med. 1994;235(5):479-485.
- Low PA. Clin Auton Res. 2008:18(suppl 1):8-13.
- Castaño A, Drachman BM, Judge D, et al. Heart Fail Rev. 2015;20(2):163-178.
- Ruberg FL, Maurer MS, Judge DP, et al. Am Heart J.
2012;164:222-228. - Olivotto I, Cecchi F, Pogessi C, et al. Circ Heart Fail.
2012;5(4):535-546. - Drazner MH. Circulation. 2011;123(3):327-334.
- Coutinho P, Martins da Silva A, Lopes Lima JL, et al. Excerpta
Medica. 1980:88-98. - Maurer MS, Hanna M, Grogan M, et al. J Am Coll Cardiol. 2016;68(2):161-172.
- Parman Y, Adams D, Obici L, et al. Curr Opin Neurol. 2016;29(suppl 1):S3-S13.
- Grogan M, Hawkins PN, Kristen AV, et al. Poster presented at: 23rd Annual Meeting of the Heart Failure Society of America (HFSA); September 13-16, 2019; Philadelphia, PA.
- Jacobson D, Tagoe C, Schwartzbard A, et al. Am J Cardiol. 2011;108(3):440-444.
- Parker MM, Damrauer SM, Rader DJ, et al. Presented at: AANEM Annual Meeting; October 10-13, 2018.
- Reilly MM, Staunton H, Harding AE, et al. J Neurol Neurosurg
Psych. 1995;59(1):45-49. - Sattianayagam PT, Hahn AF, Whelan CJ, et al. Eur Heart J. 2012;33:1120-1127.
- Adams D, Ando Y, Beirao JM, et al. J Neurol. 2021;268(6):2109-2122.
- Gillmore JD, Maurer MS, Falk RH, et al. Circulation.
2016;133(24):2404-2412. - Maurer MS, Elliott P, Comenzo R, et al. Circulation.
2017;135(14):1357-1377. - Adams D, Suhr OB, Hund E, et al. Curr Opin Neurol. 2016;29(suppl 1):S14-S26.
Small-fiber neuropathy
Cathy, 55 years old
Patient history and presentation
-
History of bilateral CTS, release surgeries in both hands
-
Progressive sensory neuropathy that began with neuropathic pain and paresthesia in feet
3 months ago and extended to lower legs -
Referred to gastroenterologist 2 years prior with alternating diarrhea and constipation, which have become more severe over the past month
-
Recent onset of paresthesia in the hands
-
Recent onset of early satiety and weight loss
-
Neurologic exam reveals: Muscle weakness in the hands and feet, impaired balance
-
Family history of sensory neuropathy
Results of clinical assessments
-
Nerve conduction study: Reduced motor and sensory responses in lower extremities
-
QSART: Reduced sweat volume in the distal legs and feet
Progressive sensory-motor and autonomic neuropathy may suggest hATTR amyloidosis.1,31,3
Consider genetic testing to help confirm a TTR variant.

Polyneuropathy and nephropathy
Mia, 48 years old
Patient history and presentation
-
Presented to neurologist 3 months prior for evaluation of numbness and distal weakness; still seeking a diagnosis
-
Returned for follow-up due to decline in ambulation over the past month; has started to use a cane when she leaves the house
-
Under the care of a nephrologist for the last
12 months for management of mild renal insufficiency and recurrent urinary tract infections (4 during the past 6 months) -
Recurrent nausea and vomiting over the past
9 months -
History of hypertension
-
Peripheral edema
Laboratory results
-
Elevated serum creatinine
-
Proteinuria
Results of clinical assessments
-
Nerve conduction study: Reduced motor and sensory responses in lower extremities
Rapidly progressive sensory-motor neuropathy with multisystem involvement may suggest hATTR amyloidosis.2,3,112,3,10
Consider genetic testing to help confirm a TTR variant.

Autonomic neuropathy symptoms, HFpEF
Sam, 60 years old
Patient history and presentation
-
Sexual dysfunction over past year
-
Alternating episodes of diarrhea and constipation
-
Recent onset of numbness in both feet
-
Reports recent bouts of dizziness when standing up
-
History of hypertension; treated with beta blocker that was stopped because of persistent hypotension
-
Dyspnea on exertion over past 6 months
-
Referred by general practitioner to cardiologist for arrhythmia and lower extremity edema
Results of clinical assessments
-
Echo: LV ejection fraction 55%; LV wall thickness 15 mm at posterior wall
-
Elevated NT-proBNP and troponin I
-
ECG: Pseudoinfarction pattern
-
CMRI: Diffuse subendocardial late gadolinium enhancement of both ventricles
-
Heart rate deep breathing test: Absent heart rate variability
Sensory and autonomic neuropathy with HFpEF may suggest hATTR amyloidosis.1,3,8
Consider genetic testing to help confirm a TTR variant.

Bilateral CTS, atrial fibrillation, intolerant to medication Charles, 66 years old
Patient history and presentation
-
History of bilateral CTS and mild sensory-motor neuropathy
-
Persistent atrial fibrillation was diagnosed
1 month prior, treated with calcium channel blocker and anticoagulants -
Returned for follow-up appointment, with new-onset orthostatic hypotension, palpitations, tachycardia, fatigue; unable to work
-
Family history of heart failure; father and aunt died at ages 62 and 64, respectively
Results of clinical assessments
-
2D Echo: LV ejection fraction 50%; thickened ventricular walls, speckled myocardium
-
ECG: High-degree AV block; low QRS voltage
-
Scintigraphy: Grade 3 myocardial uptake of 99mTc-PYP
Now is the time to rule out other types of amyloidoses. Monoclonal protein studies (serum and urine immunofixation electrophoresis, serum free light chain assay) can rule out AL amyloidosis, and genetic testing can be considered to help confirm a TTR variant.8,31
